
大家好,这里专注表观组学十余年,领跑多组学科研服务的易基因。
干旱胁迫是对农业生产产生不利影响的关键环境因素。为此,植物发展出各种响应机制(干旱逃逸、避免、耐受和回复),以通过进化增强抗旱性,这些适应机制从分子到植物水平都所不同。黄毛草莓(Fragaria nilgerrensis)是一种具有良好抗旱性的二倍体野生草莓,可为提高栽培草莓抗旱性提供有用的候选基因。到目前为止,其参与干旱胁迫相关的分子调控网络尚不清楚。
2022年10月28日,云南大学农学院乔琴教授团队与云南中医药大学张体操团队联合在《BMC Plant Biol》杂志发表题为“Integrated transcriptome and methylome analyses reveal the molecular regulation of drought stress in wild strawberry (Fragaria nilgerrensis)”的研究论文,研究通过对黄毛草莓(F. nilgerrensis)干旱胁迫处理过程中四个不同时间点的基因表达谱、全基因组DNA甲基化图谱和生理性状的综合分析,研究了F. nilgerrensis的干旱响应调控网络。

标题:Integrated transcriptome and methylome analyses reveal the molecular regulation of drought stress in wild strawberry (Fragaria nilgerrensis) 整合转录组和甲基化组分析揭示了野生草莓干旱胁迫的分子调控
时间:2022-10-28
期刊:BMC Plant Biology
影响因子:IF 5.26
技术平台:WGBS、RNA-seq、qRT-PCR分析等
项目设计:
采集同一克隆的F. nilgerrensis野生草莓作为实验材料,在温室中进行干旱处理实验,分为对照组和处理组:(i)对照组:每2天浇水一次;(ii)干旱处理组:植物从0d到12d没有浇水。在持续干旱胁迫处理期间的四个时间点收集F. nilgerrensis叶片:0d(T0,CK),4d(T4),8d(T8)和12d(T12)(图1A)。在干旱胁迫下的每个时间点的上午9点~10点之间对叶片进行取样,立即在液氮中冷冻,-80℃保存。在4个时间点获得的叶片用于DNA甲基化分析(WGBS)、转录组分析(RNA-seq)和qRT-PCR分析的材料,,实验设置三个生物学重复。
结果图形
(1)干旱胁迫下F. nilgerrensis的生理性状

图1:干旱胁迫实验材料
(A) F.nilgerrensis对干旱响应的形态变化;
(B) 在干旱处理的四个时间点检测五种生理性状。五个重复的显著差异用不同字母表示(P<0.05,方差分析)
(2)干旱胁迫下差异表达基因(DEG)的变化
转录组测序(RNA-seq)分析结果表明,差异表达基因(DEG)数量随着干旱胁迫的加剧而增加。与T0相比,在T8时间点检测到的DEG最多(5308),其中显著上调基因和下调基因数量分别为2225和3083(图2A)。值得注意的是,在T12和T8之间的DEG数量最少(100个),表明T8和T12的基因表达模式相似。使用STEM程序搜索常见的时间表达模式,发现11个重要的配置文件,其中两个宽图谱在T8时表现出上调(1549个基因)或下调(1274个基因)(图2B)。KEGG富集分析结果表明,上调基因在甘油磷脂代谢通路、MAPK信号和核糖体生物合成中富集,而下调基因在固碳作用和光合作用中富集(图2B)。这些基因可能在F.nilgerensis的干旱胁迫响应中发挥关键作用。
T8与T0时间点的DEG进行分析,GO富集分析结果表明,更多通路主要在抗旱性方面富集。上调基因主要在响应水分剥夺、氧化应激、脱落酸激活信号通路、UDP-葡糖基转移酶活性、钙调蛋白结合和活性跨膜转运蛋白活性响应中富集(图2C)。下调基因主要在光合作用、细胞壁组织或生物发生、跨膜受体蛋白激酶活性和水通道活性中富集。在T8时间点参与DEG的主要富集通路大致描绘了F.nilgerrensis抗旱性的基因参与过程。
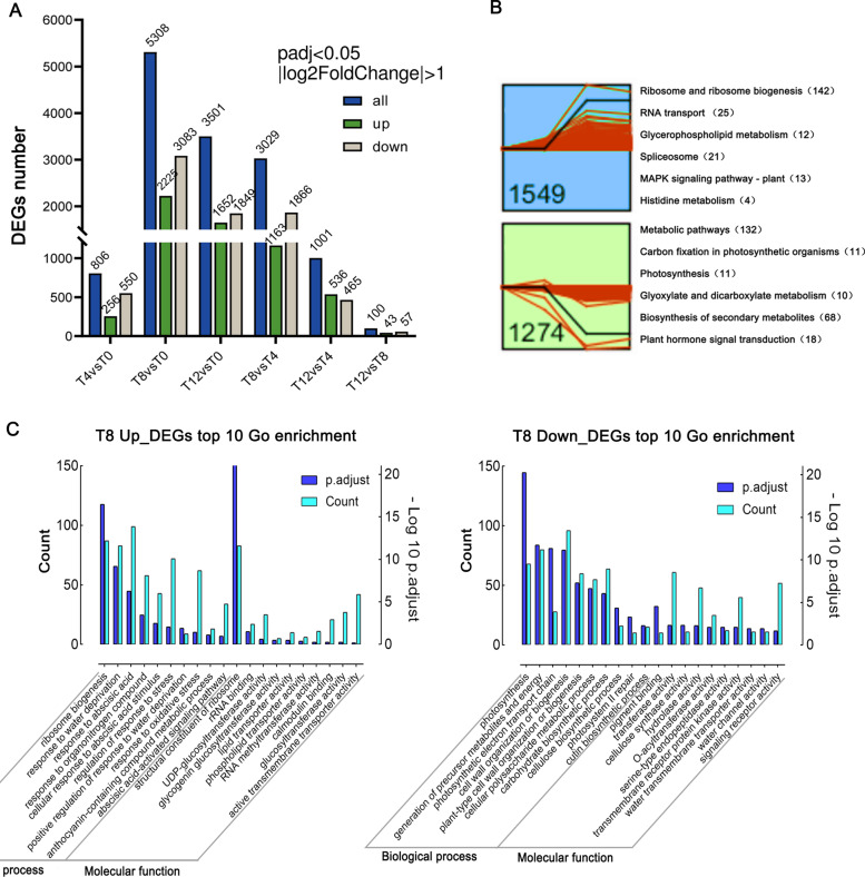
图2:差异表达基因(DEG)检测和功能注释。
(A) 每个时间点上调和下调的DEG数量;
(B) 在干旱第8天(T8)获得两个显著聚类。显示了两个簇的KEGG富集,每个通路中富集的单基因数量;
(C) T8和T0之间DEG的前10个GO富集分析
(3)干旱胁迫下F. nilgerensis的DNA甲基化图谱变化
为研究干旱胁迫下DNA甲基化水平的调控机制,研究人员进行了全基因组重亚硫酸盐测序(WGBS)分析。共12个样本生成了126Gb clean data,平均每个样本10.5Gb,测序深度>30×(基因组305.9Mb),唯一比对率为67.75%~73.13%。最低的Q20和Q30分别为96.78%和90.18%,亚硫酸盐转化率在为99.499%~99.782%,表明DNA甲基化reads信息高度可靠。
在F.nilgerensis的三个序列环境中,CG、CHG和CHH中表现出的DNA甲基化水平分别为47.69%、30.87%和10.56%(图3A)。此外, mCG、mCHG和mCHH在总mC位点中的百分比在不同时间点表现出动态变化,其中mCHH不仅占最高百分比,而且表现出最大变化(图3B)。这与之前在桑椹和陆地棉中的研究结果一致,即CHH甲基化水平随环境而动态变化,且CHH甲基化可能与干旱胁迫密切相关。在干旱胁迫期间,F. nilgerensis基因组分和重复序列的DNA甲基化水平也显示出CHH甲基化的显著变化,尤其是在启动子和重复序列中(图3C)。
此外还分析每个时间点处理组和对照(T0)之间差异甲基化区域(DMR)数,分别在T4和T8检测到大量低甲基化和高甲基化DMR(图3D)。这些DMR主要发生在CHH环境中,其中约80%位于重复元件中。不同基因组区域中每个元件的甲基化水平表明,重复元件在所有三种情况和所有时间点显示出最高的甲基化水平,其次是启动子和内含子(图3C)。由于启动子和基因体的甲基化对基因表达有不同的影响,将差异甲基化基因(DMG)分为启动子DMG和基因体DMG。结果表明,在干旱胁迫的所有时间点,F. nilgerrenis的启动子DMG均高于基因体DMG(图3D)。在启动子DMG中,所有时间点有317个低甲基化共有基因和21个高甲基化共有基因(图3E),表明这些基因的甲基化水平动态变化,可能直接或间接参与调控表达基因对干旱胁迫的调控。KEGG富集分析结果表明,这些基因主要参与植物激素信号、MAPK信号通路和泛素介导的蛋白水解通路。DMG的KEGG富集分析结果与DEG的KEGG富集分析结果大致一致。

图3:干旱胁迫下四个时间点DNA甲基化水平变化。
(A) T8时间点不同序列背景下的DNA甲基化水平变化。x轴表示甲基化水平,y轴表示总甲基化胞嘧啶位点的分数;
(B) 干旱不同时间点CG、CHG和CHH环境中mC相对比例;
(C) DNA甲基化的动态变化,包括基因组分和重复序列;
(D) 计算每个比较时间点的差异甲基化区域(DMR)及其相应基因(DMG)数量;
(E) 对照组启动子甲基化相关基因的Upset-Venn图
(4)干旱胁迫下甲基化水平与基因表达的关系
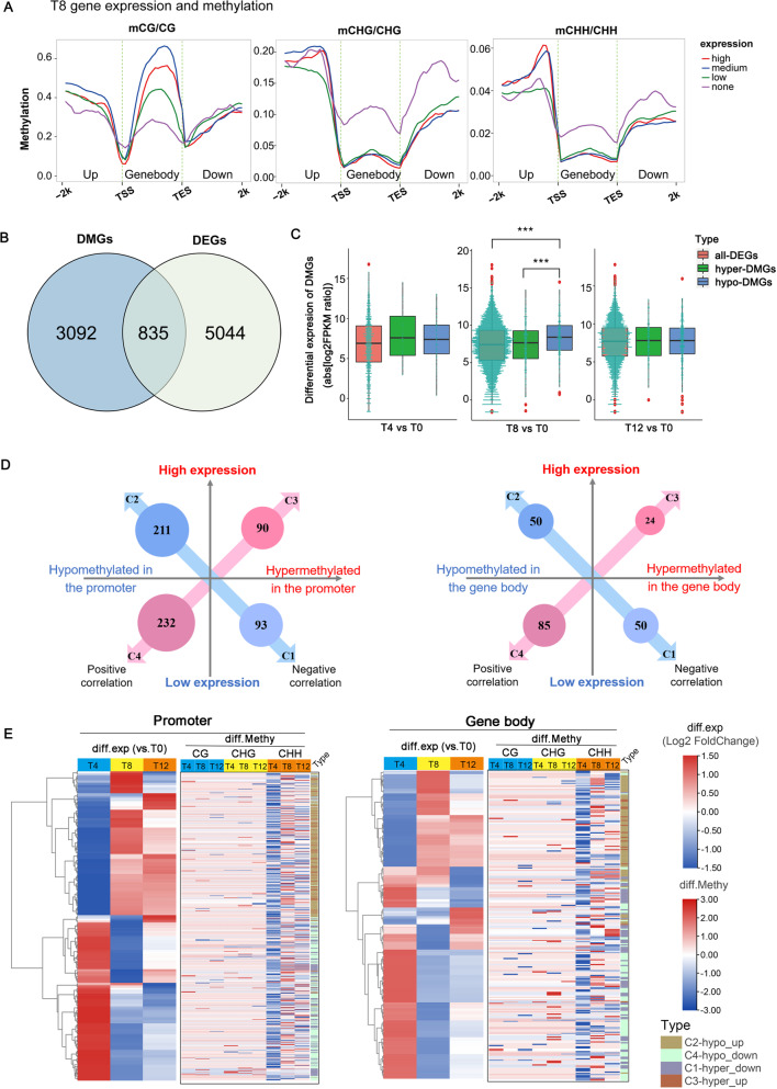
图4:甲基化组和转录组关联分析揭示干旱响应基因的表达调控。
(A) 不同表达类别的基因组分区域(基因体及其上游和下游2kb区域)的甲基化水平:低至高表达水平对应于FPKM的下/中/上四分位数,FPKM<1表示无表达(none);
(B) 共有的835个基因进行甲基化组-转录组关联分析;
(C) 所有DEG、高甲基化和低甲基化DGE基因表达水平箱线图。中间的绿松石点图表示基因数量和对应的表达水平。***P.adj< 0.001(ANOVA,Tukey_HSD);
(D,E)鉴定启动子和基因体甲基化与835个基因表达的关联。C1:高甲基化和低表达;C2:低甲基化和高表达;C3:高甲基化和高表达;C4:低甲基化和低表达。
(5)F. nilgerensis干旱响应关键基因的表征

图5:加权基因共表达网络分析(WGCNA)揭示了关键抗旱基因和抗旱模块的qRT-PCR验证。
(A)F.nilgerrensis在干旱条件下的ABA信号调控模型。红色上下箭头分别表示基因的上调和下调,“Me”表示差异甲基化;PA/DPA:相酸和二氢相酸;
(B) 模块-性状相关性。列对应由不同颜色指示的模块,行对应干旱生理特性,相交的细胞数表示相关性和P值;
(C) 高度相关的绿松石模块的相关网络。ABA信号的核心组分基因以“红色”为特征,权重以节点大小为特征,反映了与其相关的基因数量;
(D) 通过实时定量PCR验证了干旱胁迫下响应基因的表达水平。条形图表示三个生物学重复的±SD

图6:F.nilgerrensis的干旱响应调控网络。热图左侧的绿色条表示发生差异甲基化。
RuBisco:核酮糖-1,5-二磷酸羧化酶(Ribulose-1,5-bisphosphate carboxylase)
PPDK:丙酮酸,磷酸二激酶(Pyruvate, phosphate dikinase)
PEPcase:磷酸烯醇丙酮酸羧激酶(Phosphoenolpyruvate carboxykinase)
LEA:成膜晚期丰富蛋白(Late embriogenesis abundant protein )
FBPase:果糖-1,6-二磷酸酶(Fructose-1,6-bisphosphatase )
POD:过氧化物酶(Peroxide Enzyme )
ROBH:呼吸爆发氧化酶同源物(Respiratory burst oxidase homolog )
GST:谷胱甘肽转移酶(Glutathione transferase )
GPX:谷胱甘肽过氧化物酶(Glutathione peroxidase )
FER:铁蛋白(Ferritin )
TRX:硫氧还蛋白(Thioredoxin )
PIP:水通道蛋白(Aquaporins )
APX:抗坏血酸过氧化物酶(Ascorbate peroxidase )
GR:谷胱甘肽还原酶(Glutathione reductase)
结论
F. nilgerrensis是提高栽培草莓抗旱性的重要野生来源。本研究通过对DNA甲基化、转录组和生理性状的综合分析,揭示了F.nilgerrensis的干旱响应调控网络。本研究结果表明,ABA依赖性和非依赖性信号通路在F.nilgerrensis的干旱响应中发挥作用。此外,渗透调控能力和维持ROS再生与清除平衡能力对于预防代谢功能障碍至关重要,且在很大程度上决定了F.nilgerrensis的整体抗旱性。DNA甲基化和基因表达之间的相关性更为微妙,在启动子和基因体中都显示出正相关和负相关,表明DNA甲基化和基因表达之间存在多种类型的关联。综上所述,本研究为全面探索植物抗旱机制提供了模型,为育种和作物管理提供了参考。
关于易基因全基因组重亚硫酸盐测序(WGBS)技术
全基因组重亚硫酸盐甲基化测序(WGBS)可以在全基因组范围内精确的检测所有单个胞嘧啶碱基(C碱基)的甲基化水平,是DNA甲基化研究的金标准。WGBS能为基因组DNA甲基化时空特异性修饰的研究提供重要技术支持,能广泛应用在个体发育、衰老和疾病等生命过程的机制研究中,也是各物种甲基化图谱研究的首选方法。
易基因提供的全基因组甲基化测序技术通过T4-DNA连接酶,在超声波打断基因组DNA片段的两端连接接头序列,连接产物通过重亚硫酸盐处理将未甲基化修饰的胞嘧啶C转变为尿嘧啶U,进而通过接头序列介导的 PCR 技术将尿嘧啶U转变为胸腺嘧啶T。
应用方向:
WGBS广泛用于各种物种,要求全基因组扫描(不错过关键位点)
Ø 全基因组甲基化图谱课题
Ø 标志物筛选课题
Ø 小规模研究课题
技术优势:
Ø 应用范围广:适用于所有参考基因组已知物种的甲基化研究;
Ø 全基因组覆盖:最大限度地获取完整的全基因组甲基化信息,精确绘制甲基化图谱;
Ø 单碱基分辨率:可精确分析每一个C碱基的甲基化状态。
易基因科技提供全面的DNA甲基化研究整体解决方案,详询易基因:0755-28317900。
参考文献:
Cao Q, Huang L, Li J, Qu P, Tao P, Crabbe MJC, Zhang T, Qiao Q. Integrated transcriptome and methylome analyses reveal the molecular regulation of drought stress in wild strawberry (Fragaria nilgerrensis). BMC Plant Biol. 2022 Dec 28;22(1):613.
相关阅读:
疾病研究:DNA甲基化与转录组分析联合揭示吸烟免疫相关疾病的表观遗传机制
疾病研究:DNA甲基化和转录组学特征在高浆卵巢癌复发和耐药过程中高度保守
动物发育:DNA甲基化组与转录组综合分析绒山羊胚胎期毛囊发育的调控机制


